

L'électrophorèse
Histoire:
L'origine de cette technique a été imaginée par S.E. Linder et H. Picton en 1892. Ils se sont inspirés des études de Hermann Von Helmholtz menées sur l'électro-osmose. Celui-ci constate qu'il est possible, sous un champ électrique, de déplacer des particules chargées vers le pôle de signe opposé à leur charge.En 1937, Arne Wilhelm Kaurin Tiselius, met au point la première électrophorèse: l'électrophorèse libre. Cette technique lui a permis de séparer les protéines du serum sanguin en appliquant un champ électrique. La solution est placée dans un tube en U de section carrée afin de réaliser des mesures optiques au travers du tube.
Il a pu ainsi obtenir sur le pôle + des protéines de charge très négative comme l'abumine et sur le pôle - des protéines de charge plus positive comme les globulines.
Cette technique ne permet toutefois pas de séparer totalement les protéines. Il est néanmoins possible de mettre en évidence les frontières formées par des méthodes optiques comme la fluorescence, l'absorption des UV ou l'indice de réfraction.
En 1939, P. König et D Von Klobusitzky ont séparé avec succès les composants du venin de serpent en élaborant la technique d'électrophorèse sur papier.
En 1952, Pierre Grabar élabore, en collaboration avec C.A. Williams, une méthode connue sous le nom d'analyse immuno-électrophorétique, qui permet d'analyser de manière précise des mélanges très complexes d'antigènes. Dès la première application de cette méthode à l'analyse du sérum sanguin humain, il parvient à déceler dans le sérum plus de 30 constituants indépendants, alors que l'électrophorèse en veine liquide ou sur papier ne permettait d'isoler que 5 ou 6 groupes de protéines. La méthode est rapidement utilisée dans de nombreux laboratoires médicaux pour des besoins de diagnostiques.
En 1955, O. Smithies met au point la technique d'électrophorèse en gel d'amidon.
En 1957, Joachim Kohn sépare les différents phénotypes de l'hémoglobines en élaborant la technique d'électrophorèse sur membrane d'acétate de cellulose.
En 1969, Beber et Osborn introduisent l'agent dénaturant SDS (Sodium Dodecyl Sulfate) pour séparer les différentes sous-unités protéiques.
Principe:
L'électrophorèse est une technique permettant de déplacer des ions (molécules ayant perdu leur neutralité électrique) sous l'effet d'un champ électrique. Ceux-ci migrent vers leur électrode respective: les anions (chargés négativement) migrent vers l'anode (potentiel positif) et les cations (chargés positivement) migrent vers la cathode (potentiel négatif). En ce qui concerne les molécules non chargées, il n'existe pas de migration. Du fait de leurs caractéristiques propres et des conditions de l'électrophorèse, la vitesse de migration et la distance parcourue dans la matrice par ces ions différent. Cela permet ainsi de les séparer.Avantages:
L'électrophorèse permet de traiter simultanément plusieurs échantillons en même temps.
La séparation est fine.
On peut déterminer 2 types d'électrophorèses:
L'électrophorèse dite "libre" en veine liquide (mise au point par Tisélius en 1937).
L'électrophorèse de zone sur support.
L'électrophorèse en veine liquide.
La migration dans ce type d'électrophorèse s'effectue au sein d'un liquide de composition déterminée soumis à un champ électrique de courant continu (cf. Histoire).Avantage:
Permet une bonne détermination des mobilités électrophorétiques.
Inconvénients:
Appareillage couteux.
La mise en oeuvre et longue et délicate.
Les particules ne se séparent pas complètement mais il se forme des frontières mise en évidence par des méthodes optiques telles que l'absoption ultra-violette pour les protéines.
L'électrophorèse de zone.
Ce type d'électrophorèse utilise un support poreux pour stabiliser la phase liquide. Le support doit être homogène, poreux et inerte. Différents types de support peuvent être utilisés:Support en papier ou acétate (la migration des mélanges s'effectue à la surface de celui-ci).
Support en polyacrylamide ou agarose (la migration des mélanges s'effectue à l'intérieur même du support).
Avantages:
Les fractions séparées migrent comme des "zones" individuelles.
La taille des mailles ("pores") pour les supports en gel peut influencer la vitesse de migration (ralentir le déplacement des grosses molécules).
2 types de montage peuvent être mis en place:
Montage horizontal:
Utilisé pour les supports en acétate de cellulose ou en en papier. Le support se présente sous forme de longue et étroite bande.
Les extrémités du support plongent dans un tampon d'électrode, créant une mince couche d'eau à sa surface.
Montage vertical:
Utilisé pour les supports en gel polyacrylamide ou, plus rarement d'agarose . Le gel est souvent préparé peu avant usage en le coulant entre 2 plaques de verre. Durant la gélification on aura pris soin de faire des puits où on déposera les échantillons. Chaque extrémité du gel est mis en contact avec un tampon contenant des électrolytes qui permettra la propagation d'un courant dans le gel.
Electrophorèse sur gel de polyacrylamide ou PAGE (Poly-Acrylamide Gel Electrophoresis):
Cette technique est très utilisée en immunologie et dans l'étude des protéines car elle permet, après la séparation des différentes protéines, leur transfert sur une membrane de nitrocellulose ou de polyfluorure de vinylidène afin d'être identifier par le biais d'anticorps spécifiques.
Cette technique est également utilisée pour le séquençage de l'ADN (les méthodes traditionnelles de Maxam et Gilbert ou de Sanger permettent de séquencer à la paire de bases près).
La matrice de cette électrophorèse:
Le gel de polyacrylamide est constitué d'acrylamide (extrêmement neurotoxique par ingestion ou contact avec la peau) qui est l'unité de base et de bisacrylamide qui est l'agent portant. En faisant varier les taux de ces 2 substances on obtient différents maillages et donc différentes densités de gel.
La polymérisation est réalisée grace à l'ajout de 2 réactifs: le TEMED (N,N,N',N' tetra-méthyl-èthylènediamine) et l'ammonium persulfate qui deviennent des anions hyperactifs en présence de lumière.
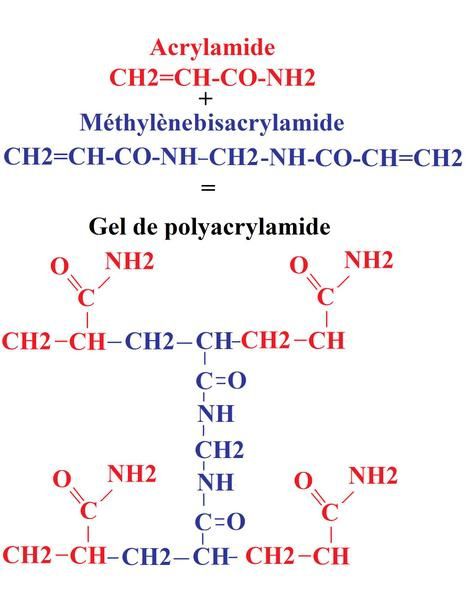
La densité typique d'un gel de séparation peut être à 6%; 8%; 10%; 12% ou 15%.
Plus le gel est dense meilleure sera la séparation des protéines.
Des niveaux de densité plus faibles du gel permettent de séparer des protéines de poids moléculaires proches.
La densité d'un gel de concentration (stacking gel) est à 5%. Ce gel est coulé en haut du gel de séparation. Il permet à l'échantillon une entrée homogène dans le gel de séparation. On utilise un "peigne" afin de créer des "puits" individuels pouvant accueillir chaque échantillon.
Le SDS-PAGE:
Une variante de cette technique consiste à utiliser du SDS (Sodium Dodécylsulfate) qui est un détergent anionique fort. Il a la propriété de défaire la structure spatiale en se fixant sur les protéines et de les charger de la même façon permettant ainsi de les séparer uniquement en fonction de leur masse moléculaire.
Les protéines sont dites dénaturées: elles ont perdu leur structure tridimentionnelle native.
Avant de procéder à la dénaturation des protéines avec du SDS, on utilise un agent réducteur, le b mercaptoéthanol qui réduit les ponts disulfures des proteines les rendant ainsi sous forme monomérique.
****
Protocole expérimental:préparation des échantillons:
Les échantillons vont être traités dans du tampon de dénaturation . Celui-ci sera préparé au dernier moment.
tampon de dénaturation se compose:
de tampon Tris Hcl 0,125 mL pH 6,8
d'eau distillée;
de SDS à 4%
de b mercaptoéthanol à 10%;
de bleu de bromophénol à 0,2%
de glycérol pur
50 microlitres d'échantillons sont mélangés avec 50 microlitres de tampon dénaturant.
L'ensemble est ensuite chauffé pendant 4 minutes afin de favoriser la dénaturation des protéines.
Le matériel d'électrophorèse se compose:
des plaques
des pinces
le joint d'étanchéité
Le peigne
les espaceurs
On nettoie les plaques avant utilisation à l'aide d'eau (éventuellement savonneuse) puis avec de l'alcool à 70%. On essuie les plaques avec un papier, sans laisser les fibres sur les faces où sera coulé le gel.
On pose le joint d'étanchéité sur la plaque à bords rond.
Puis, mise en place des espaceurs et de la deuxième plaque en verre. L'ensemble est ensuite consolidé à l'aide des pinces.
Préparation de 30 ml de gel de séparation:
7,50 mL d'acrylamide bis acrylamide
11,25 mL de tampon Tris/Hcl pH8,8
0,30 mL de SDS à 10%
10,95 mL d'eau
pour la polymérisation on ajoute
30 microlitres de TEMED
et après agitation, on ajoute
300 microlitres de persulfate d'ammonium à 20%
La préparation est aussitôt coulée entre les 2 plaques à l'aide d'une pipette. Afin d'éviter la présence de bulle dans le gel, on incline le montage lors du remplissage. Le gel est ensuite recouvert d'eau distillée afin d'obtenir une surface parfaitement horizontale.
Préparation de 10 ml de gel de concentration:
1,25 mL d'acrylamide bis acrylamide
1,25 mL de tampon Tris/Hcl pH6,8
0,10 mL de SDS à 10%
7,4 mL d'eau
pour la polymérisation on ajoute
40 microlitres de TEMED
et après agitation, on ajoute
100 microlitres de persulfate d'ammonium à 20%
Retirer l'eau distillée au dessus du gel de séparation puis remplir le montage comme précèdemment jusqu'au niveau supérieur des plaques de verre. Le peigne est alors mis en place pour former des puits dans le gel de concentration.
Préparation d'un litre de tampon de migration:
12,5 mL de Tris 25mM
14,4 g de glycine
977,5 mL d'eau distillée
10 mL SDS 10%
Le peigne est retiré puis les plaques et les cuves de migration sont placées sur le support . Les cuves sont remplies avec le tampon de migration en commençant par la partie supérieure puis inférieure. Dépot de 50 microlitres d'échantillon protéique dénaturé dans chaque puit à l'aide d'une seringue et selon la technique sous-marine.
Les électrodes de la cuve sont reliées au générateur. La migration se fait à 30 mA jusqu'à ce que le bleu de bromophénol atteigne le bord inférieur des plaques.
Révélation:
Le gel est retiré des plaques puis placé sur un agitateur et coloré pendant 30 minutes dans une solution de coloration composée de bleu de coomassie à 2,5%,d'isopropanol à 20%, d'acide acétique à 20% et d'eau à 57,5% puis décoloré pendant 3 fois 30 minutes avec une solution décolorante composée de 45% d'isopropanol, de 10% d'acide acétique et de 45% d'eau.
Partager cet article



Pour être informé des derniers articles, inscrivez vous :
/image%2F1216766%2F20200114%2Fob_052c5a_microscope.jpg)
/http%3A%2F%2Fe.educlever.com%2Fimg%2F1%2F8%2F2%2F5%2F18259.gif%23width%3D%26height%3D)
/image%2F1216766%2F20171016%2Fob_9bccc1_structuredesphages1-1.jpg)
/image%2F1216766%2F20141127%2Fob_c55855_hematimetre-nachet-1.jpg)
Commenter cet article
M
B
M
